Investigating the Bird Flu Virus
This example shows how to calculate Ka/Ks ratios for eight genes in the H5N1 and H2N3 virus genomes, and perform a phylogenetic analysis on the HA gene from H5N1 virus isolated from chickens across Africa and Asia. For the phylogenetic analysis, you will reconstruct a neighbor-joining tree and create a 3-D plot of sequence distances using multidimensional scaling. Finally, you will map the geographic locations where each HA sequence was found on a regional map. Sequences used in this example were selected from the bird flu case study on the Computational Genomics Website [1]. Note: The final section in this example requires the Mapping Toolbox™.
Introduction
There are three types of influenza virus: Type A, B and C. All influenza genomes are comprised of eight segments or genes that code for polymerase B2 (PB2), polymerase B1 (PB1), polymerase A (PA), hemagglutinin (HA), nucleoprotein (NP), neuraminidase (NA), matrix (M1), and non-structural (NS1) proteins. Note: Type C virus has hemagglutinin-esterase (HE), a homolog to HA.
Of the three types of influenza, Type A has the potential to be the most devastating. It affects birds (its natural reservoir), humans and other mammals and has been the major cause of global influenza epidemics. Type B affects only humans causing local epidemics, and Type C does not tend to cause serious illness.
Type A influenzas are further classified into different subtypes according to variations in the amino acid sequences of HA (H1-16) and NA (N1-9) proteins. Both proteins are located on the outside of the virus. HA attaches the virus to the host cell then aids in the process of the virus being fused in to the cell. NA clips the newly created virus from the host cell so it can move on to a healthy new cell. Difference in amino acid composition within a protein and recombination of the various HA and NA proteins contribute to Type A influenzas' ability to jump host species (i.e. bird to humans) and wide range of severity. Many new drugs are being designed to target HA and NA proteins [2,3,4].
In 1997, H5N1 subtype of the avian influenza virus, a Type A influenza virus, made an unexpected jump to humans in Hong Kong causing the deaths of six people. To control the rapidly spreading disease, all poultry in Hong Kong was destroyed. Sequence analysis of the H5N1 virus is shown here [2,4].
Calculate Ka/Ks Ratio For Each H5N1 Gene
An investigation of the Ka/Ks ratios for each gene segment of the H5N1 virus will provide some insight into how each is changing over time. Ka/Ks is the ratio of non-synonymous changes to synonymous in a sequence. For a more detailed explanation of Ka/Ks ratios, see Analyzing Synonymous and Nonsynonymous Substitution Rates. To calculate Ka/Ks, you need a copy of the gene from two time points. You can use H5N1 virus isolated from chickens in Hong Kong in 1997 and 2001. For comparison, you can include H2N3 virus isolated from mallard ducks in Alberta in 1977 and 1985 [1].
For the purpose of this example, sequence data is provided in four MATLAB® structures that were created by genbankread.
Load H5N1 and H2N3 sequence data.
load('birdflu.mat','chicken1997','chicken2001','mallard1977','mallard1985')
Data in public repositories is frequently curated and updated. You can retrieve the up-to-date datasets by using the getgenbank function. Note that if data has indeed changed, the results of this example might be slightly different when you use up-to-date datasets.
chicken1997 = arrayfun(@(x)getgenbank(x{:}),{chicken1997.Accession});
chicken2001 = arrayfun(@(x)getgenbank(x{:}),{chicken2001.Accession});
mallard1977 = arrayfun(@(x)getgenbank(x{:}),{mallard1977.Accession});
mallard1985 = arrayfun(@(x)getgenbank(x{:}),{mallard1985.Accession});
You can extract just the coding portion of the nucleotide sequences using the featureparse function. The featureparse function returns a structure with fields containing information from the Features section in a GenBank file including with a Sequence field that contains just the coding sequence.
for ii = 1:numel(chicken1997) ntSeq97{ii} = featureparse(chicken1997(ii),'feature','cds','sequence',true); ntSeq01{ii} = featureparse(chicken2001(ii),'feature','cds','sequence',true); ntSeq77{ii} = featureparse(mallard1977(ii),'feature','cds','sequence',true); ntSeq85{ii} = featureparse(mallard1985(ii),'feature','cds','sequence',true); end ntSeq97{1}
ans =
struct with fields:
Location: '<1..>2273'
Indices: [1 2273]
UnknownFeatureBoundaries: 1
gene: 'PB2'
codon_start: '1'
product: 'PB2 protein'
protein_id: 'AAF02361.1'
db_xref: 'GI:6048850'
translation: 'RIKELRDLMSQSRTREILTKTTVDHMAIIKKYTSGRQEKNPALRMKWMMAMKYPITADKRIMEMIPERNEQGQTLWSKTNDAGSDRVMVSPLAVTWWNRNGPTTSTVHYPKVYKTYFEKVERLKHGTFGPVHFRNQVKIRRRVDMNPGHADLSAKEAQDVIMEVVFPNEVGARILTSESQLTITKEKREELKNCNISPLMVAYMLERELVRKTRFLPVAGGTSSVYIEVLHLTQGTCWEQMYTPGGEVRNDDVDQSLIIAARNIVRRATVSADPLASLLEMCHSTQIGGVRMVDILKQNPTEEQAVDICKAAMGLRISSSFSFGGFTFKRTKGFSVKREEEVLTGNLQTLKIQVHEGYEEFTMVGRRATAILRKATRRMIQLIVSGRDEQSIAEAIIVAMVFSQEDCMIKAVRGDLNFVNRANQRLNPMHQLLRHFQKDAKVLFQNWGIEPIDNVMGMIGILPDMTPSTEMSLRGVRVSKMGVDEYSSTERVVVSIDRFLRVRDQQGNVLLSPEEVSETQGMEKLTITYSSSMMWEINGPESVLVNTYQWIIRNWETVKIQWSQEPTMLYNKMEFEPFQSLVPKAARSQYSGFVRTLFQQMRDVLGTFDTVQIIKLLPFAAAPPEQSRMQFSSLTVNVRGSGMRILVRGNSPAFNYNKTTKRLTILGKDAGAITEDPDEGAAGVESAVLRGFLILGKEDKRYGPALSINELSNLTKGEKANVLIGQGDVVLVMKRKRDSSILTDSQTATKRIRMAIN'
Sequence: 'agaataaaagaactaagagatttgatgtcgcaatctcgcacacgcgagatactgacaaaaaccactgtggatcatatggccataattaagaagtacacatcaggaagacaggagaagaaccccgctcttagaatgaaatggatgatggcgatgaaatacccgatcacagctgacaaaagaataatggagatgatccctgaaaggaatgagcaaggtcaaactctttggagcaaaacaaatgacgctggatcagacagggtaatggtatcacctctggctgtaacgtggtggaacagaaatggaccaacaacaagtacagtccattatccaaaggtgtataaaacctactttgaaaaggttgaaagattaaaacacggaacctttggccctgttcatttccggaatcaagtcaaaatacgccgcagggttgacatgaaccctggccatgcagatctcagcgctaaagaagcacaagatgtcatcatggaggtcgttttcccaaatgaagttggagccaggatattgacatcagagtcacagctgacaataacaaaggaaaagagggaggaactcaagaattgtaatatttctcctttaatggtggcatatatgttggaaagagaattggttcgcaagaccagattcctaccagtggctggtgggacaagcagcgtatatatagaagtattgcatttgacccaaggaacctgctgggagcagatgtacacaccaggaggggaggtaagaaatgatgatgttgaccaaagtttaatcattgctgctaggaacattgtcaggagagcaacagtatcagcagacccattggcttcactcttggagatgtgccatagcacacaaattggcggagtaagaatggtagacatccttaaacaaaacccaacagaagagcaagctgtagatatatgcaaggcagcaatgggtttaagaatcagctcatccttcagctttggagggttcactttcaaaagaacaaaggggttttctgtcaaaagagaggaagaagtgcttacaggcaacctccaaacattgaagatacaagtacatgaaggatatgaggaattcacaatggttggacgaagagcaacagccattctaagaaaagcaaccagaaggatgatccaactgatagtcagcgggagggacgagcaatcaattgctgaggcaattattgtagcaatggtgttctcacaagaagattgcatgataaaggcagtccgaggtgatttgaatttcgtaaacagagcaaatcaacgactgaaccccatgcaccaactcctgagacacttccaaaaggatgcaaaggtgctgtttcaaaactggggaattgaaccaatcgacaatgtcatggggatgattggaatattgcctgacatgacccccagcacggaaatgtcactaagaggagtgagagttagtaaaatgggggtggatgaatattctagcactgaaagagtggtcgtgagcattgaccgtttcttaagggtccgagatcagcaaggaaatgtactcctatcccctgaagaagttagtgagacacagggaatggaaaagttgacgataacttattcatcgtctatgatgtgggaaattaacggcccagaatcagttctagttaacacataccaatggatcattaggaattgggagactgtaaagatccaatggtcccaagaacccaccatgctatacaataagatggagtttgaaccatttcaatctttagtaccaaaggctgccagaagccaatatagtggatttgtgagaacgctattccagcagatgcgtgatgttttgggaacatttgacactgttcaaataatcaaactactaccatttgcagcagccccacctgaacagagtaggatgcaattttcttctctgactgtgaatgtgaggggatcaggaatgagaatacttgtgagaggtaactcccctgcgtttaactacaacaagacaactaagaggcttacaatacttgggaaggacgcaggtgcaattacagaggacccagatgaaggagcagcaggggtagagtctgcagtattgagagggtttctaattctcggcaaagaagacaaaagatatggaccagcattgagcatcaatgaactgagcaatcttacgaaaggggagaaagctaatgtattgatagggcaaggagacgtggtgttggtaatgaaacggaaacgggactctagcatacttactgacagccagacagcgaccaaaagaattcggatggccatcaatta'
Visual inspection of the sequence structures revealed some of the genes have splice variants represented in the GenBank files. Because this analysis is only on PB2, PB1, PA, HA, NP, NA, M1, and NS1 genes, you need to remove any splice variants.
Remove splice variants from 1997 H5N1
ntSeq97{7}(1) = [];% M2
ntSeq97{8}(1) = [];% NS2
Remove splice variants from 1977 H2N3
ntSeq77{2}(2) = [];% PB1-F2
ntSeq77{7}(1) = [];% M2
ntSeq77{8}(1) = [];% NS2
Remove splice variants from 1985 H2N3
ntSeq85{2}(2) = [];% PB1-F2
ntSeq85{7}(1) = [];% M2
ntSeq85{8}(1) = [];% NS2
You need to align the nucleotide sequences to calculate the Ka/Ks ratio. Align protein sequences for each gene (available in the 'translation' field) using nwalign function, then insert gaps into nucleotide sequence using seqinsertgaps. Use the function dnds to calculate non-synonymous and synonymous substitution rates for each of the eight genes in the virus genomes. If you are interested in seeing the sequence alignments, set the 'verbose' option to true when using dnds.
Influenza gene names
proteins = {'PB2','PB1','PA','HA','NP','NA','M1','NS1'};
H5N1 Virus
for ii = 1:numel(ntSeq97) [sc,align] = nwalign(ntSeq97{ii}.translation,ntSeq01{ii}.translation,'alpha','aa'); ch97seq = seqinsertgaps(ntSeq97{ii}.Sequence,align(1,:)); ch01seq = seqinsertgaps(ntSeq01{ii}.Sequence,align(3,:)); [dn,ds] = dnds(ch97seq,ch01seq); H5N1.(proteins{ii}) = dn/ds; end
H2N3 Virus
for ii = 1:numel(ntSeq77) [sc,align] = nwalign(ntSeq77{ii}.translation,ntSeq85{ii}.translation,'alpha','aa'); ch77seq = seqinsertgaps(ntSeq77{ii}.Sequence,align(1,:)); ch85seq = seqinsertgaps(ntSeq85{ii}.Sequence,align(3,:)); [dn,ds] = dnds(ch77seq,ch85seq); H2N3.(proteins{ii}) = dn/ds; end H5N1 H2N3
H5N1 =
struct with fields:
PB2: 0.0226
PB1: 0.0240
PA: 0.0307
HA: 0.0943
NP: 0.0517
NA: 0.1015
M1: 0.0460
NS1: 0.3010
H2N3 =
struct with fields:
PB2: 0.0048
PB1: 0.0021
PA: 0.0089
HA: 0.0395
NP: 0.0071
NA: 0.0559
M1: 0
NS1: 0.1954
Note: Ka/Ks ratio results may vary from those shown on [1] due to sequence splice variants.
Visualize Ka/Ks ratios in 3-D bar graph.
H5N1rates = cellfun(@(x)(H5N1.(x)),proteins);
H2N3rates = cellfun(@(x)(H2N3.(x)),proteins);
bar3([H2N3rates' H5N1rates']);
ax = gca;
ax.XTickLabel = {'H2N3','H5N1'};
ax.YTickLabel = proteins;
zlabel('Ka/Ks');
view(-115,16);
title('Ka/Ks Ratios for H5N1 (Chicken) and H2N3 (Mallard) Viruses');
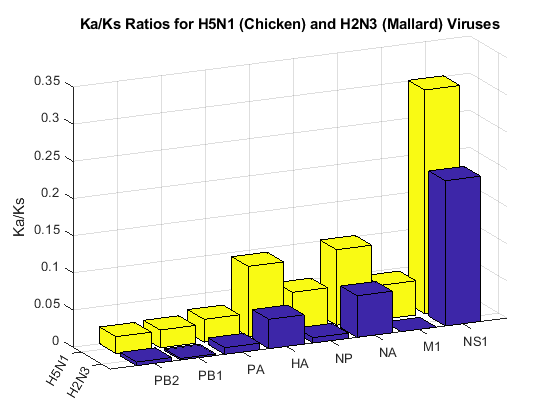
NS1, HA and NA have larger non-synonymous to synonymous ratios compared to the other genes in both H5N1 and H2N3. Protein sequence changes to these genes have been attributed to an increase in H5N1 pathogenicity. In particular, changes to the HA gene may provide the virus the ability to transfer into others species beside birds [2,3].
Perform a Phylogenetic Analysis of the HA Protein
The H5N1 virus attaches to cells in the gastrointestinal tract of birds and the respiratory tract of humans. Changes to the HA protein, which helps bind the virus to a healthy cell and facilitates its incorporation into the cell, are what allow the virus to affect different organs in the same and different species. This may provide it the ability to jump from birds to humans [2,3]. You can perform a phylogenetic analysis of the HA protein from H5N1 virus isolated from chickens at different times (years) in different regions of Asia and Africa to investigate their relationship to each other.
Load HA amino acid sequence data from 16 regions/times from the MAT-file provided birdflu.mat or retrieve the up-to-date sequence data from the NCBI repository using the getgenpept function.
load('birdflu.mat','HA')
HA = arrayfun(@(x)getgenpept(x{:}),{HA.Accession});
Create a new structure array containing fields corresponding to amino acid sequence (Sequence) and source information (Header). You can extract source information from the HA using featureparse then parse with regexp.
for ii = 1:numel(HA) source = featureparse(HA(ii),'feature','source'); strain = regexp(source.strain,'A/[Cc]hicken/(\w+\s*\w*).*/(\d+)','tokens'); proteinHA(ii).Header = sprintf('%s_%s',char(strain{1}(1)),char(strain{1}(2))); proteinHA(ii).Sequence = HA(ii).Sequence; end proteinHA(1)
ans =
struct with fields:
Header: 'Nigeria_2006'
Sequence: 'mekivllfaivslvksdqicigyhannsteqvdtimeknvtvthaqdilekthngklcdldgvkplilrdcsvagwllgnpmcdeflnvpewsyivekinpandlcypgnfndyeelkhllsrinhfekiqiipksswsdheassgvssacpyqgrssffrnvvwlikkdnayptikrsynntnqedllvlwgihhpndaaeqtrlyqnpttyisvgtstlnqrlvpkiatrskvngqsgrmeffwtilkpndainfesngnfiapenaykivkkgdstimkseleygncntkcqtpigainssmpfhnihpltigecpkyvksnrlvlatglrnspqgerrrkkrglfgaiagfieggwqgmvdgwygyhhsneqgsgyaadkestqkaidgvtnkvnsiidkmntqfeavgrefnnlerrienlnkkmedgfldvwtynaellvlmenertldfhdsnvknlydkvrlqlrdnakelgngcfefyhrcdnecmesvrngtydypqyseearlkreeisgvklesigtyqilsiystvasslalaimvaglslwmcsngslqcrici'
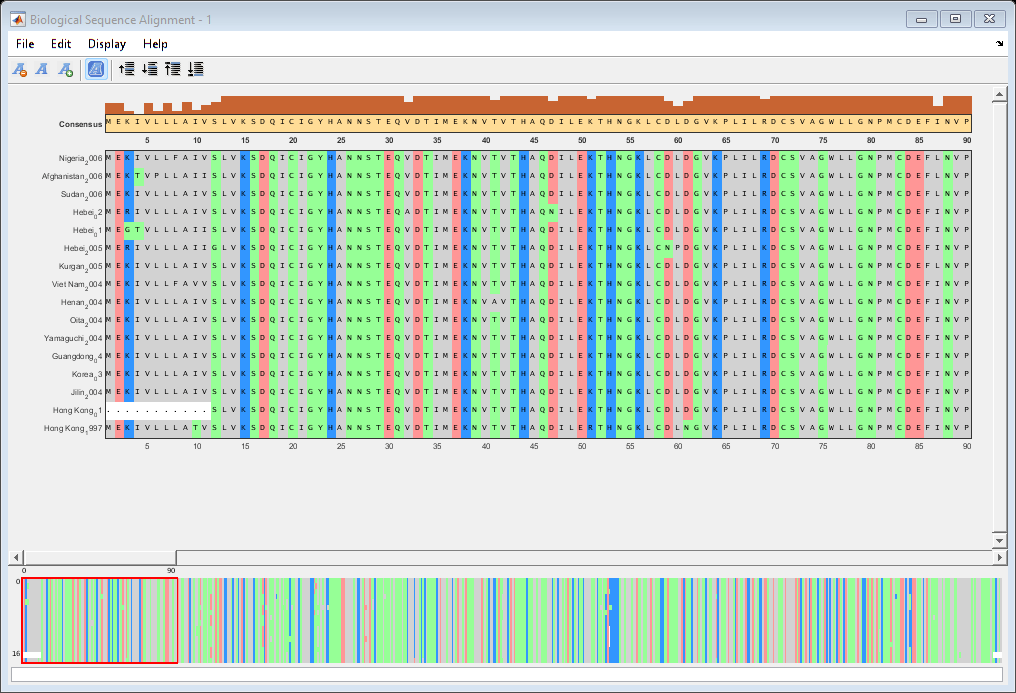
Align the HA amino acid sequences using multialign and visualize the alignment with seqalignviewer.
alignHA = multialign(proteinHA); seqalignviewer(alignHA);
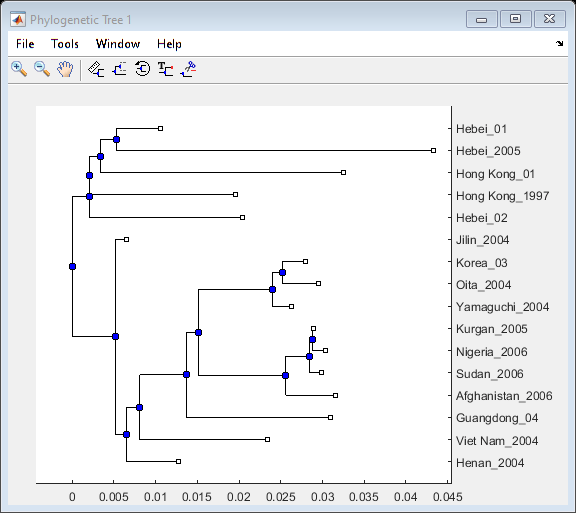
Calculate the distances between sequences using seqpdist with the Jukes-Cantor method. Use seqneighjoin to reconstruct a phylogenetic tree using the neighbor-joining method. Seqneighjoin returns a phytree object.
distHA = seqpdist(alignHA,'method','Jukes-Cantor','alpha','aa'); HA_NJtree = seqneighjoin(distHA,'equivar',alignHA);
Use the view method associated with phytree objects to open the tree in the Phylogenetic Tree Tool.
view(HA_NJtree);
Visualize Sequence Distances with Multidimensional Scaling (MDS)
Another way to visualize the relationship between sequences is to use multidimensional scaling (MDS) with the distances calculated for the phylogenetic tree. This functionality is provided by the cmdscale function in Statistics and Machine Learning Toolbox™.
[Y,eigvals] = cmdscale(distHA);
You can use the eigenvalues returned by cmdscale to help guide your decision of whether to use the first two or three dimensions in your plot.
sigVecs = [1:3;eigvals(1:3)';eigvals(1:3)'/max(abs(eigvals))]; report = ['Dimension Eigenvalues Normalized' ... sprintf('\n %d\t %1.4f %1.4f',sigVecs)]; display(report);
report =
'Dimension Eigenvalues Normalized
1 0.0062 1.0000
2 0.0028 0.4462
3 0.0014 0.2209'
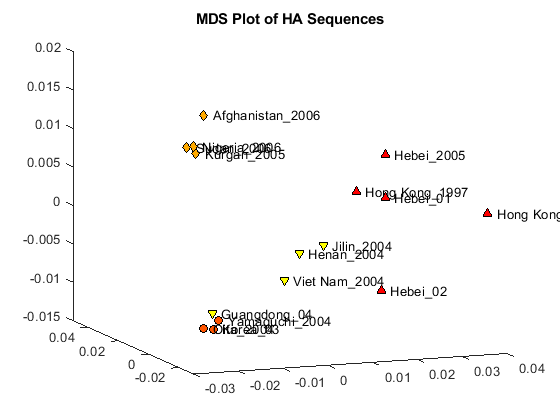
The first two dimensions represent a large portion of the data, but the third still contains information that might help resolve clusters in the sequence data. You can create a three dimensional scatter plot using plot3 function.
locations = {proteinHA(:).Header};
figure
plot3(Y(:,1),Y(:,2),Y(:,3),'ok');
text(Y(:,1)+0.002,Y(:,2),Y(:,3)+0.001,locations,'interpreter','no');
title('MDS Plot of HA Sequences');
view(-21,12);
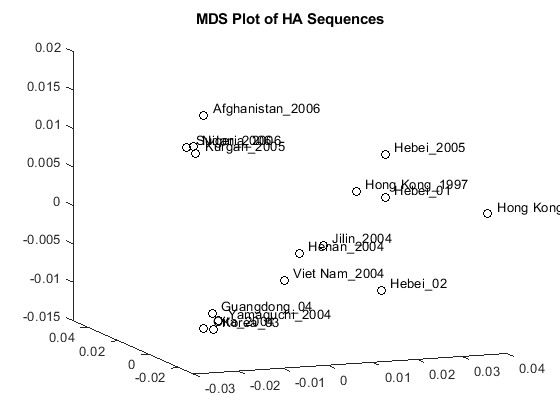
Clusters appear to correspond to groupings in the phylogenetic tree. Find the sequences belonging to each cluster using the subtree method of phytree. One of subtree's required inputs is the node number (number of leaves + number of branches), which will be the new subtree's root node. For your example, the cluster containing Hebei and Hong Kong in the MDS plot is equivalent to the subtree whose root node is Branch 14, which is Node 30 (16 leaves + 14 branches).
cluster1 = get(subtree(HA_NJtree,30),'LeafNames'); cluster2 = get(subtree(HA_NJtree,21),'LeafNames'); cluster3 = get(subtree(HA_NJtree,19),'LeafNames');
Get an index for the sequences belonging to each cluster.
[cl1,cl1_ind] = intersect(locations,cluster1);
[cl2,cl2_ind] = intersect(locations,cluster2);
[cl3,cl3_ind] = intersect(locations,cluster3);
[cl4,cl4_ind] = setdiff(locations,{cl1{:} cl2{:} cl3{:}});
Change the color and marker symbols on the MDS plot to correspond to each cluster.
h = plot3(Y(cl1_ind,1),Y(cl1_ind,2),Y(cl1_ind,3),'^',... Y(cl2_ind,1),Y(cl2_ind,2),Y(cl2_ind,3),'o',... Y(cl3_ind,1),Y(cl3_ind,2),Y(cl3_ind,3),'d',... Y(cl4_ind,1),Y(cl4_ind,2),Y(cl4_ind,3),'v'); numClusters = 4; col = autumn(numClusters); for i = 1:numClusters h(i).MarkerFaceColor = col(i,:); end set(h(:),'MarkerEdgeColor','k'); text(Y(:,1)+0.002,Y(:,2),Y(:,3),locations,'interpreter','no'); title('MDS Plot of HA Sequences'); view(-21,12);
For more detailed information on using Ka/Ks ratios, phylogenetics and MDS for sequence analysis, see Cristianini and Hahn [5].
Optional: Display Geographic Regions of the H5N1 Virus on a Map of Africa and Asia
If you have Mapping toolbox, you can run the following code to generate a figure that shows the geographic locations of the virus.
Create a geostruct structure, regionHA, that contains the geographic information for each feature, or sequence, to be displayed. A geostruct is required to have Geometry, Lat, and Lon fields that specify the feature type, latitude and longitude. This information is used by mapping functions in Mapping Toolbox to display geospatial data.
[regionHA(1:16).Geometry] = deal('Point'); [regionHA(:).Lat] = deal(9.10, 34.31, 15.31, 39.00, 39.00, 39.00, 55.26,... 15.56, 34.00, 33.14, 34.20, 23.00, 37.35, 44.00,... 22.11, 22.11); [regionHA(:).Lon] = deal(7.10, 69.08, 32.35, 116.00, 116.00, 116.00,... 65.18, 105.48, 114.00, 131.36, 131.40, 113.00,... 127.00, 127.00, 114.14, 114.14);
A geostruct can also have attribute fields that contain additional information about each feature. Add attribute fields Name and Cluster to the regionHA structure. The Cluster field contains the sequence's cluster number, which you will use to identify the sequences' cluster membership.
[regionHA(:).Name] = deal(proteinHA.Header); [regionHA(cl1_ind).Cluster] = deal(1); [regionHA(cl2_ind).Cluster] = deal(2); [regionHA(cl3_ind).Cluster] = deal(3); [regionHA(cl4_ind).Cluster] = deal(4);
Create a structure using the makesymbolspec function, which will contain marker and color specifications for each marker to be displayed on the map. You will pass this structure to the geoshow function. Symbol markers and colors are set to correspond with the clusters in MDS plot.
clusterSymbols = makesymbolspec('Point',... {'Cluster',1,'Marker', '^'},... {'Cluster',2,'Marker', 'o'},... {'Cluster',3,'Marker', 'd'},... {'Cluster',4,'Marker', 'v'},... {'Cluster',[1 4],'MarkerFaceColor',autumn(4)},... {'Default','MarkerSize', 6},... {'Default','MarkerEdgeColor','k'});
Load the mapping information and use the geoshow function to plot virus locations on a map.
load coastines load topo figure fig = gcf; worldmap([-45 85],[0 160]) setm(gca,'mapprojection','robinson',... 'plabellocation',30,'mlabelparallel',-45,'mlabellocation',30) geoshow(coastlat,coastlon) geoshow(topo, topolegend, 'DisplayType', 'texturemap') demcmap(topo) brighten(.60) geoshow(regionHA,'SymbolSpec',clusterSymbols); title('Geographic Locations of HA Sequence in Africa and Asia')
close all
References
[1] https://computationalgenomics.blogs.bristol.ac.uk/case_studies/birdflu_demo
[2] Laver, W.G., Bischofberger, N. and Webster, R.G., "Disarming Flu Viruses", Scientific American, 280(1):78-87, 1999.
[3] Suzuki, Y. and Masatoshi, N., "Origin and Evolution of Influenza Virus Hemagglutinin Genes", Molecular Biology and Evolution, 19(4):501-9, 2002.
[4] Gambaryan, A., et al., "Evolution of the receptor binding phenotype of influenza A(H5) viruses", Virology, 344(2):432-8, 2006.
[5] Cristianini, N. and Hahn, M.W., "Introduction to Computational Genomics: A Case Studies Approach", Cambridge University Press, 2007.
[6] Google Earth images were acquired using Google Earth Pro. For more information about Google Earth and Google Earth Pro, visit http://earth.google.com/
You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)